Next: Structural and Dynamic Properties Up: Multiscale Simulations of Graphene Previous: Multiscale Simulations of Graphene Contents
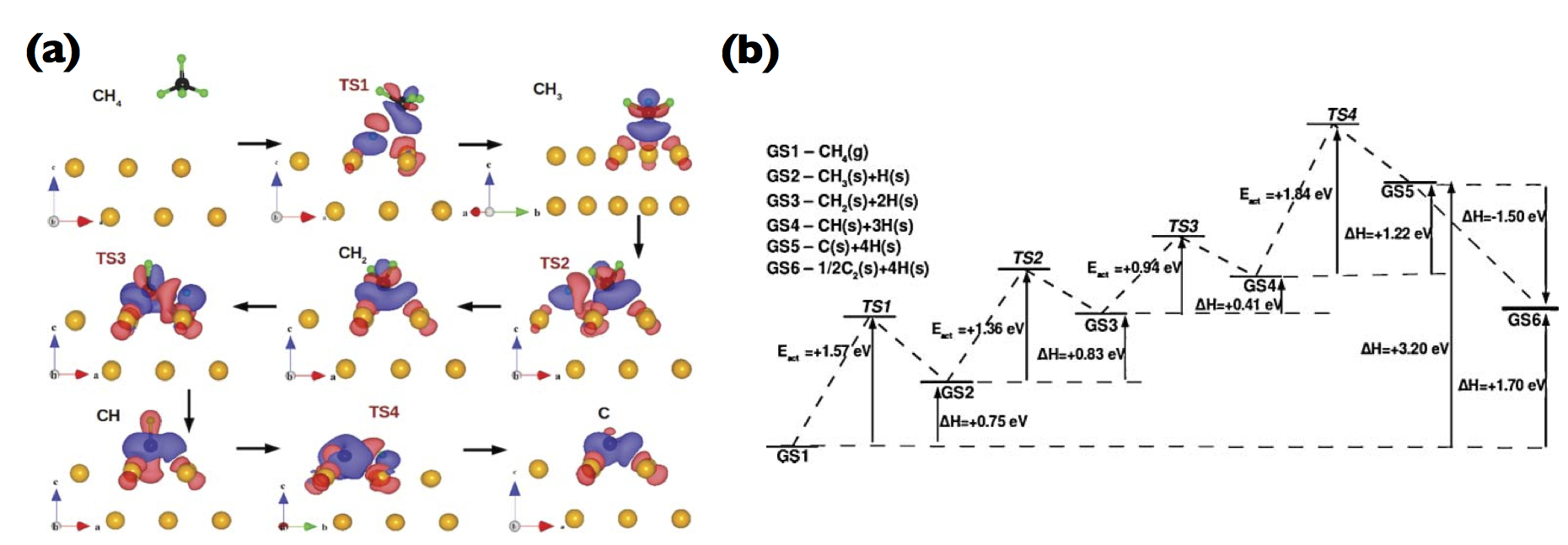
![[*]](crossref.png) a displays the electron density difference [IMAGE png] along the minimum energy pathway (MEP) of sequential dehydrogenation. From Fig. a it is evident to observe significant degrees of charge transfer between the adsorbates and the Cu atoms along the reaction path, and the existence of Cu atoms can reduce the electron density between carbon and hydrogen atoms, which weakens C-H bonds. Figure b displays the activation barriers for sequential [IMAGE png] decomposition over this Cu surface, and our calculations revealed that the reaction barriers are much lower than those in the gas phase. Hence, our MEP analysis revealed the role of Cu substrate as the catalytic material for graphene growth. All the dehydrogenation reactions are endothermic, except for carbon dimer ([IMAGE png]) formation, which is, therefore, the most critical step for subsequent graphene growth, in particular, on Cu (111) surface. This work has been published in the Journal of Chemical Physics (DOI:10.1063/1.3624524).
a displays the electron density difference [IMAGE png] along the minimum energy pathway (MEP) of sequential dehydrogenation. From Fig. a it is evident to observe significant degrees of charge transfer between the adsorbates and the Cu atoms along the reaction path, and the existence of Cu atoms can reduce the electron density between carbon and hydrogen atoms, which weakens C-H bonds. Figure b displays the activation barriers for sequential [IMAGE png] decomposition over this Cu surface, and our calculations revealed that the reaction barriers are much lower than those in the gas phase. Hence, our MEP analysis revealed the role of Cu substrate as the catalytic material for graphene growth. All the dehydrogenation reactions are endothermic, except for carbon dimer ([IMAGE png]) formation, which is, therefore, the most critical step for subsequent graphene growth, in particular, on Cu (111) surface. This work has been published in the Journal of Chemical Physics (DOI:10.1063/1.3624524).
 |
barbarossapao 2015-09-16
![[IMAGE png]](img12.png){kind=link}